Inputs/Outputs¶
The Alexandria Sheet¶
To instruct the workflow, you must create a comma-separated-value (csv) sample sheet called the Alexandria Sheet which contains sample names, paths to sequencing data on the workspace bucket, and any metadata you wish to include on Alexandria.
Formatting your Alexandria Sheet for FASTQ files¶
For processing a sequencing directory full of BCL files, see the below section instead.
Write your Alexandria Sheet in a text editor or a spreadsheet manipulation program such as Microsoft Excel and save your file as a comma-separated value (.csv) file. The Alexandria Sheet must have column headers and contains the following in whatever order:
Important
- (REQUIRED) the ‘Sample’ column; the sample/array names that must prefix the respective .fastq or .fastq.gz files. Any preexisting count matrices must be named as
<sample>_dge.txt.gzand located in the bucket at<output_path>/dropseq/<sample>/<sample>_dge.txt.gz. - (RECOMMENDED) both ‘R1_Path’ and ‘R2_Path’ columns; the paths to .fastq/.fastq.gz files on the bucket. Alternatively, see the section on the fastq_directory parameter.
- (OPTIONAL) Other metadata columns that will be appended to the alexandria_metadata.txt (tab-delimited) file generated after running Cumulus. Column headers must match exactly the names of attributes found in the Alexandria Metadata Convention. Labels outside of this convention will be supported in the future.
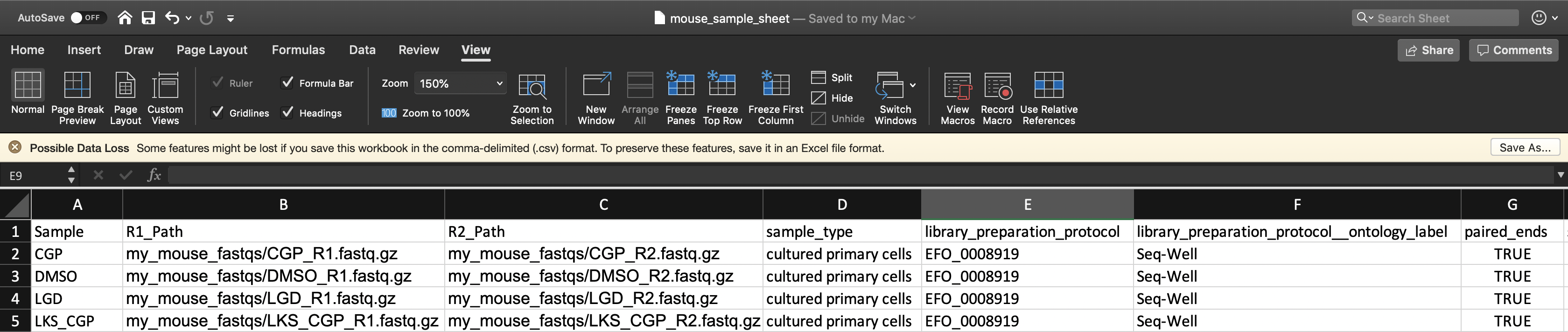
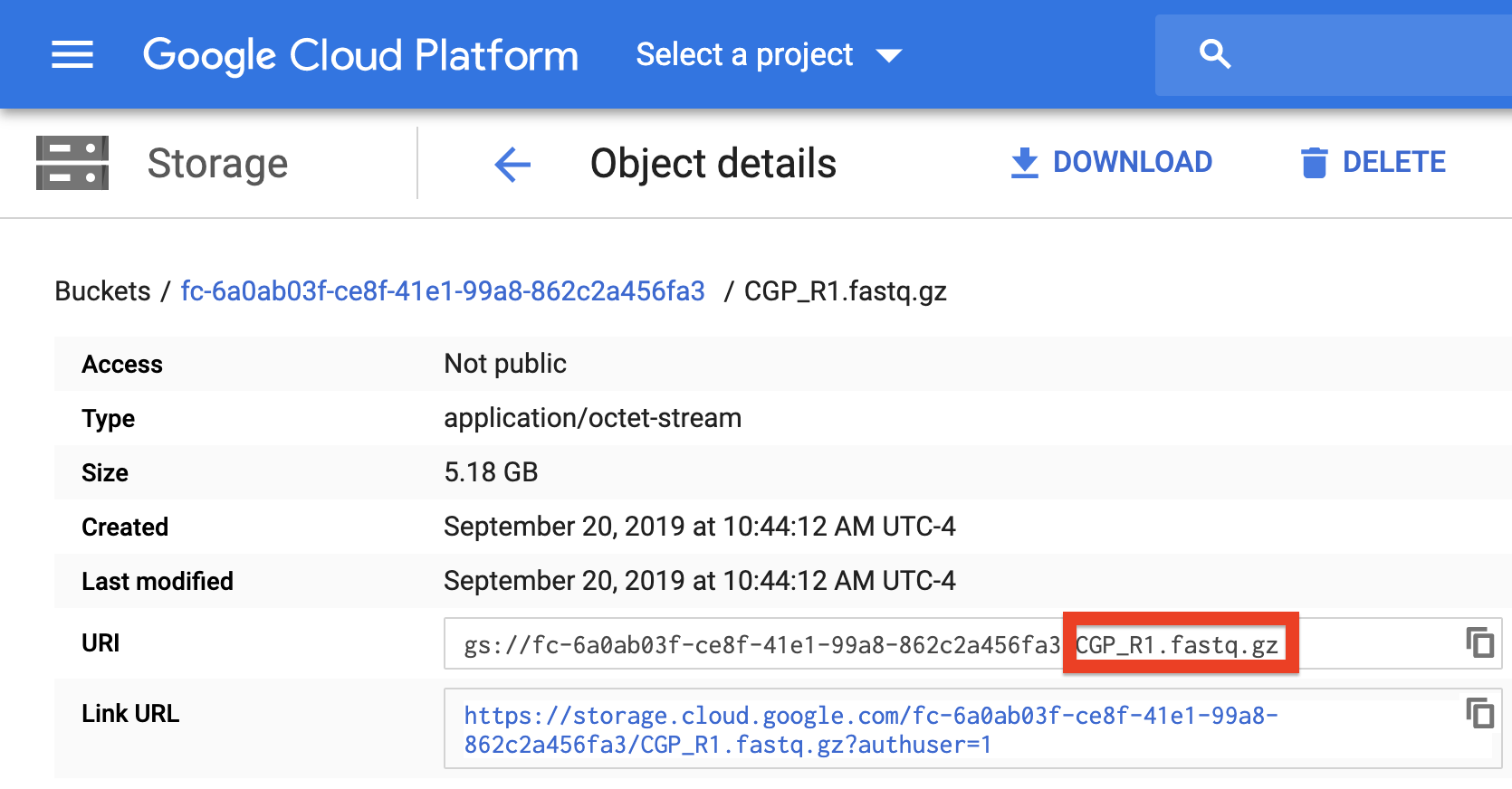
To verify that the paths you listed in the file are correct, you can navigate to your Google bucket and locate your sequence data files. Click on each file to view its URI (gsURL), which should resemble the format gs://<bucket ID>/path/to/file.fastq.gz in the case of gzip-compressed FASTQ files (regular FASTQ files are fine too). The locations you should enter in the path columns of your Alexandria Sheet can be the entire URI or all of the characters following the bucket ID and trailing slash, in this case path/to/file.fastq.gz.
Formatting your Alexandria Sheet for bcl2fastq¶
Due to legal requirements, the default Docker image for bcl2fastq workflow made by the Cumulus Team is privately available for use by Broad Institute affiliates. Affiliates must create a Docker account using their broadinstitute.org email address, download Docker Desktop, and log in through typing docker login in their terminal. Only then can you launch the bcl2fastq workflow on Alexandria or Terra.
If you are not an affiliate, you can either create and reference your own Docker image or you can download the Bcl2Fastq software here and run it locally on a computer with plenty of disk space. If you chose the latter, once you get your FASTQs you can see the above section for writing a csv to process them.
Write your Alexandria Sheet in a text editor or a spreadsheet manipulation program such as Microsoft Excel and save your file as a comma-separated value (.csv) file. The Alexandria Sheet must have column headers and contains the following in whatever order:
Important
- (REQUIRED) the ‘Sample’ column, the sample/array names that are found in each sequencing directory’s sample sheet. Any pre-existing count matrices must have the sample names prefix each .txt.gz file.
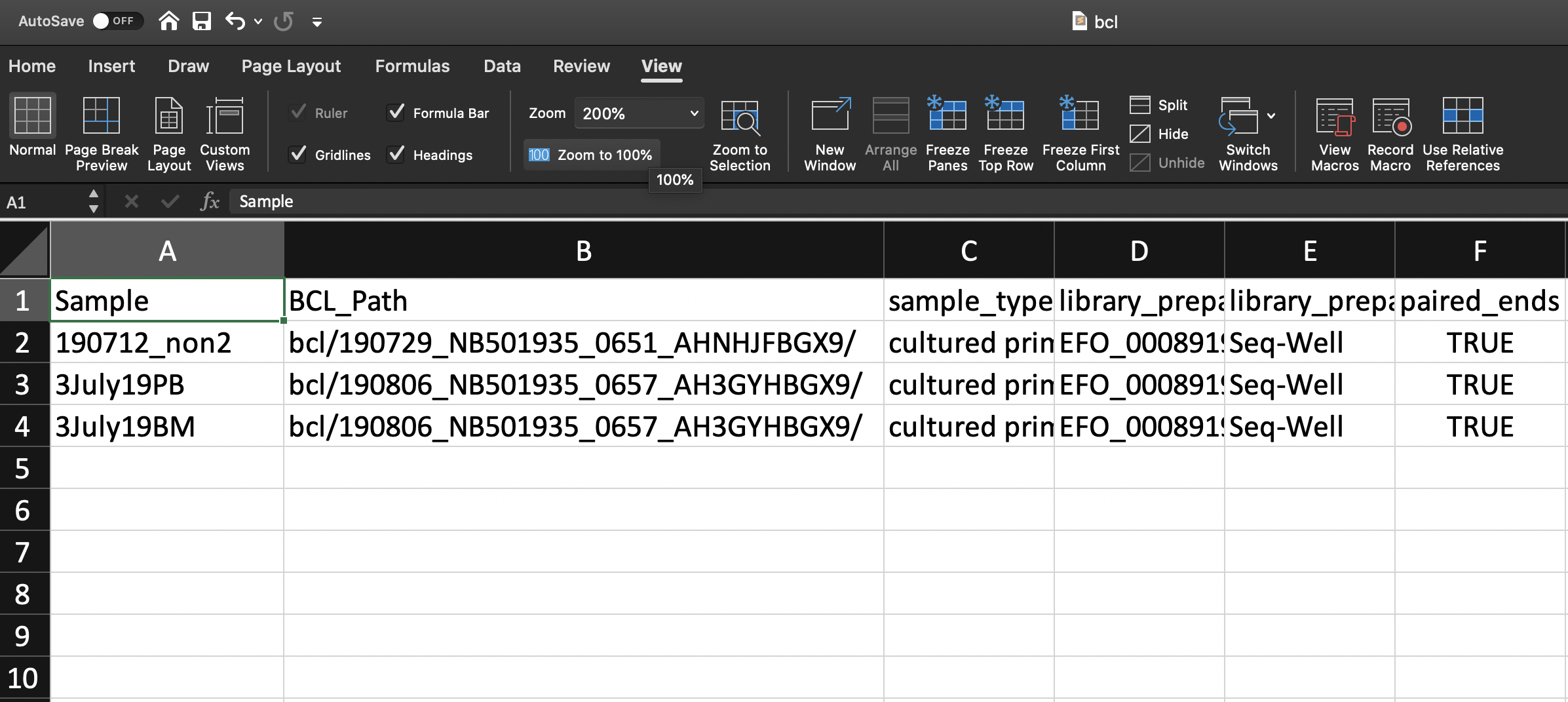
- (REQUIRED) ‘BCL_Path’ column, the paths to the sequencing run directories on the bucket. Ex:
path/to/191231_NB501935_0679_AHVY52BGXB/ - (OPTIONAL) The ‘SS_Path’ column; Recommended if you have BCL_Path and your sample sheets are not located within the root of the sequencing run directory. Paths to the sequencing run directories’
SampleSheet.csvfiles on the bucket. If blank, will check inside the corresponding BCL_Path forSampleSheet.csv. - (OPTIONAL) Other metadata columns that will be appended to the alexandria_metadata.txt file generated after running Cumulus. Column headers must match exactly the names of attributes found in the Alexandria Metadata Convention. Labels outside of this convention will be supported in the future.
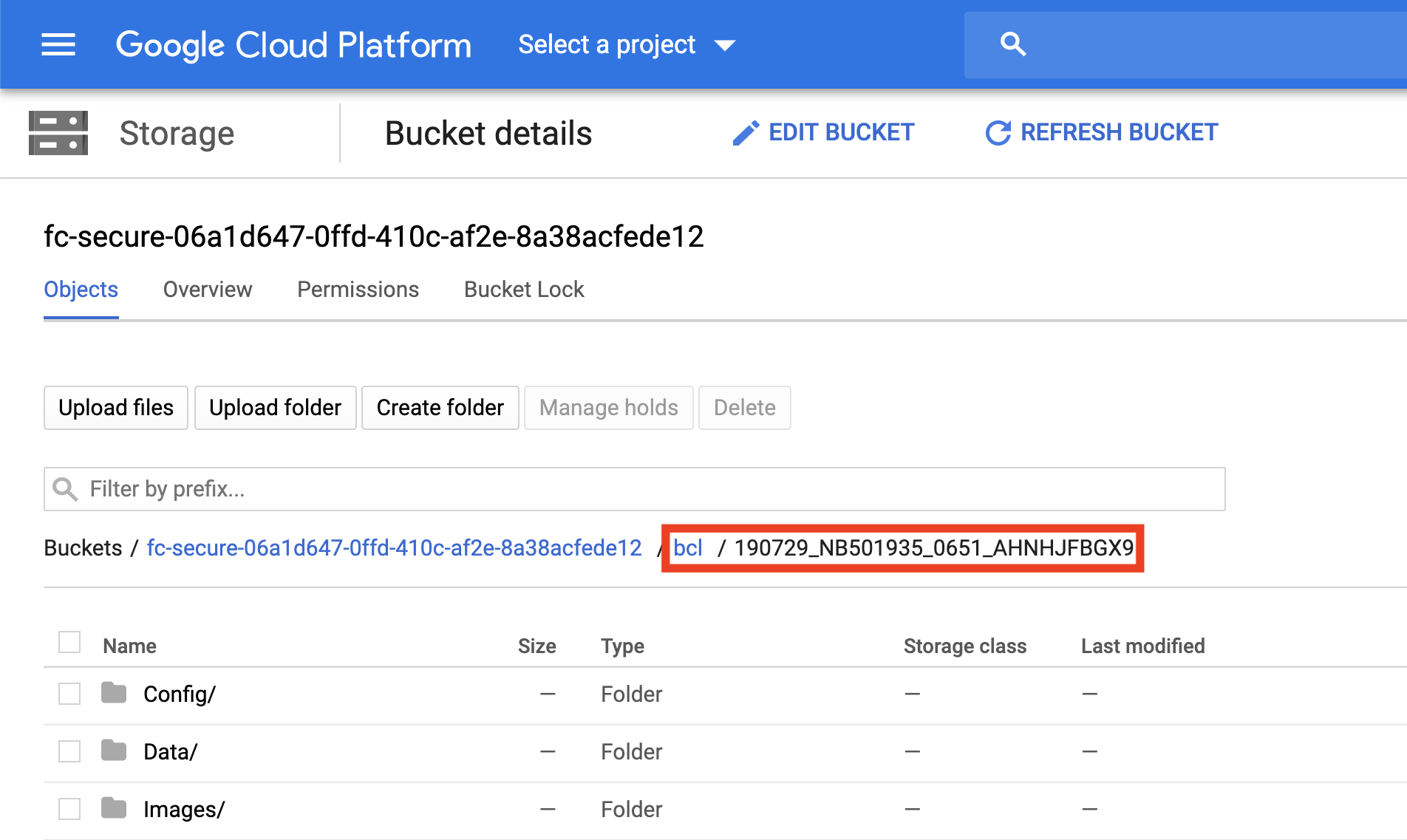
To verify that the paths you listed in the file are correct, you must navigate to your Google bucket and locate your sequence data files. Click on each file to view its URI (gsURL), which should resemble the format gs://<bucket ID>/path/to/sequencing_run_directory/ in the case of sequencing run directories. The locations you should enter in the path columns of your Alexandria Sheet should be all of the characters following the bucket ID and trailing slash, in this case path/to/sequencing_run_directory.
Understanding the fastq_directory parameter¶
The use of this variable is not essential and is only meant for quick and convenient CSV writing when you are submitting FASTQs.
Refer to the above spreadsheet example. There are four samples which each have two FASTQ reads. All FASTQ files are found in a folder located at the root of the bucket called mouse_fastqs. Since they are all located in the same directory, one could set mouse_fastqs as the fastq_directory and no longer need to have R1_Path and R2_Path columns.
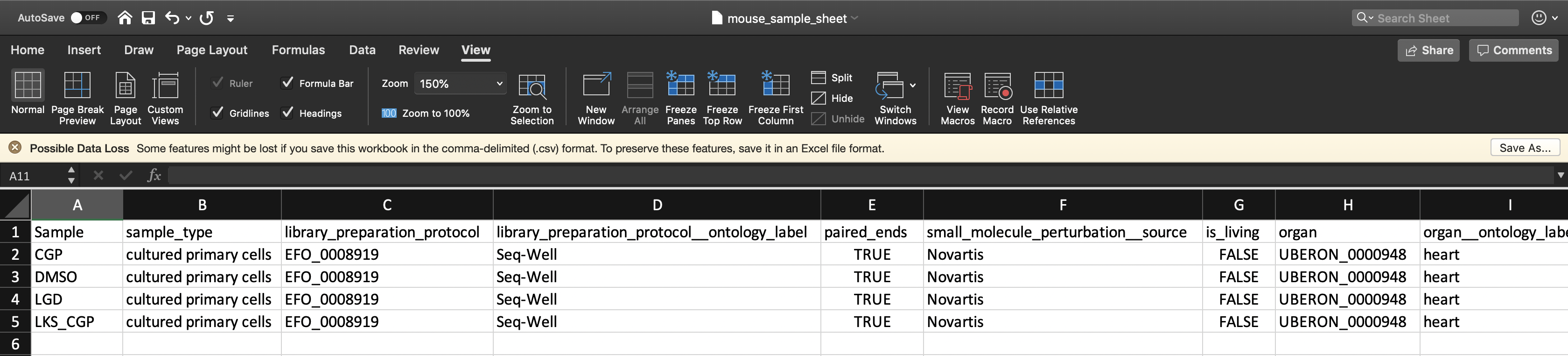
Furthermore, if the user has R1_Path and R2_Path columns but leaves spreadsheet cells left blank, the pipeline will search in the fastq_directory for the corresponding sample.
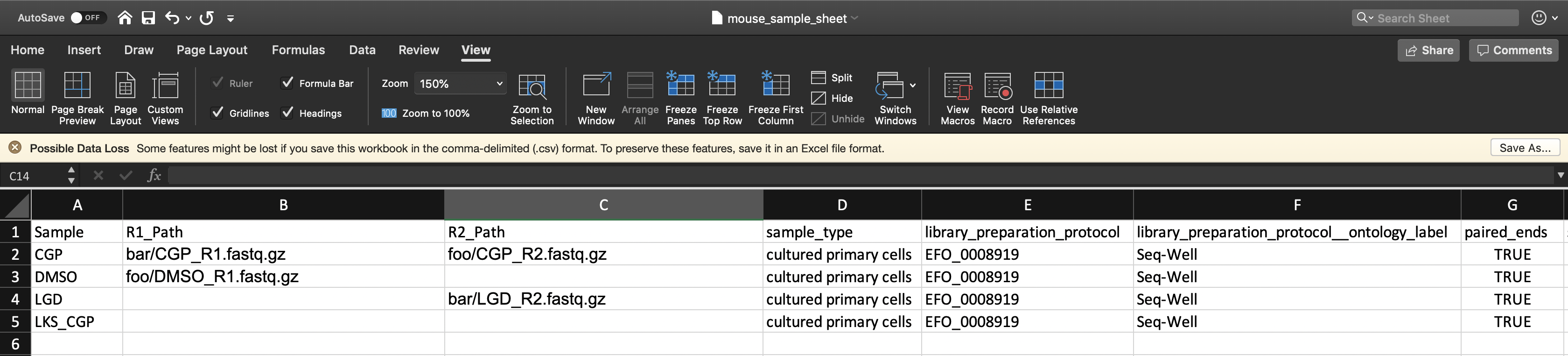
Here the pipeline will search the gs://[bucket ID]/mouse_fastqs directory for any spreadsheet cells left blank; DMSO_R2.fastq.gz, LGD_R1.fastq.gz, LKS_CGP_R1.fastq.gz, and LKS_CGP_R2.fastq.gz. The specific pattern the pipeline searches for is <Sample Name>*_<R1 or R2>*.fastq(.gz).
Inputs of the dropseq_cumulus workflow¶
Basic usage¶
| Variable | Description |
|---|---|
| bucket | gsURL of the workspace bucket to which you have permissions, ex: gs://fc-e0000000-0000-0000-0000-000000000000/. This value is not exposed on Alexandria and is locked to the workspace bucket. |
| output_path | Path to bucket folder where outputs (count matrices, metadata files, etc.) will be stored. All folders in this path will be created if they do not exist. Ex: Entering data/20200326/ stores Drop-Seq Tools outputs at gs://<bucket>/data/20200326/dropseq/ and Cumulus outputs at gs://<bucket>/data/20200326/cumulus/ |
| input_csv_file | Sample sheet (comma-separated value file) uploaded in the miscellaneous tab of this study’s Upload/Edit Study Data page. **Formatting must adhere to the criteria!** |
| reference | Genome for alignment. Supported options: hg19, GRCh38, mm10, or mmul_8.0.1. See here for building a custom genome for Drop-Seq Tools. |
| run_dropseq | Yes: run Drop-seq pipeline (sequence alignment and QC). Sequencing data must be uploaded to the Google bucket associated with this study. |
| is_bcl | Yes: bcl2fastq will be run to convert all of your BCL directories to fastq.gz. No: all of your data is already of fastq.gz type. |
| fastq_directory, default = ‘’ | Sequence data directory name for sequence uploaded to the SCP study google bucket. Ex: Enter data/mouse_fastqs for folder mouse_fastqs located at gs://study bucket ID/data/mouse_fastqs/ If not applicable, list paths in the Alexandria Sheet. |
| run_cumulus | Yes: run Cumulus (generate metadata, cluster files, coordinate files for data exploration in Alexandria). If run_cumulus Yes and run_dropseq No: each digital gene expression matrix, <sample>_dge.txt.gz, must be located within the bucket at <output_path>/dropseq/<sample>/<sample>_dge.txt.gz. |
Advanced usage¶
| Variable | Description |
|---|---|
cumulus_output_prefix, default = sco |
Prefix for Cumulus files to distinguish them from files from different Cumulus jobs. |
preemptible, default = 2 |
Number of attempts using a preemptible virtual machine before requesting a higher-cost, non-preemptible instance. See Google Cloud documentation. |
zones, default = us-east1-d us-west1-a us-west1-b |
The ordered list of Google Zone preferences for requesting a Google machine to run the pipeline. See Google Cloud documentation page. |
alexandria_docker, default = shaleklab/alexandria:0.2 |
Full address of the shaleklab/alexandria Docker image to use. |
dropseq_registry, default = cumulusprod |
Registry of the Drop-Seq Tools Docker image. Default Drop-Seq Tools image registry. |
dropseq_tools_version, default = 2.3.0 |
Image tag of the Drop-Seq Tools Docker image. Default Drop-Seq Tools image tags. |
bcl2fastq_registry, default = gcr.io/broad-cumulus |
Registry of the bcl2fastq Docker image. Default is privately hosted for Broad Institute members only, see this page to create your own. |
| bcl2fastq_version, default = ``2.20.0.422` | Image tag of the bcl2fasdtq Docker image. Default is privately hosted for Broad Institute members only, see this page to create your own. |
cumulus_registry, default = cumulusprod |
Registry of the Cumulus Docker image. Default Cumulus image registry. |
cumulus_version, default = 0.15.0 |
Image tag of the Cumulus Docker image that corresponds to the version of Cumulus. Default Cumulus image tags. |
Optional inputs exposed on Terra¶
See the Drop-Seq Pipeline workflow documentation.
Caution
dropEst does not account for strandedness and therefore its usage is not recommended.
See the Cumulus workflow documentation.
Outputs of the dropseq_cumulus workflow¶
When running the Drop-Seq pipeline and/or Cumulus through dropseq_cumulus, the workflow yields the same outputs as its component workflows. You can see those documentations in the section above.
Explicitly, dropseq_cumulus presents the Single-Cell Portal with the alexandria metadata file (alexandria_metadata.txt), the dense expression matrix (ends with scp.expr.txt), and the coordinate file (ends with scp.X_fitsne.coords.txt).